2018年3月29日,国际学术期刊《Human Gene Therapy》在线发表中南大学生命科学学院医学遗传学研究中心梁德生课题组在SMA基因编辑治疗研究的最新进展,文章题为“Seamless genetic conversion of SMN2 to SMN1 via CRISPR/Cpf1 and single-stranded oligodeoxynucleotides in spinal muscular atrophy patient-specific iPSCs”,博士研究生周妙金为第一作者,梁德生和邬玲仟教授为共同通讯作者。
脊髓性肌肉萎缩症(spinal muscular atrophy, SMA)是一类由于脊髓前角运动神经元变性导致的进行性骨骼肌无力和萎缩的一组疾病,是最常见的致死性常染色体隐性遗传病之一,发病率为1/6000~1/10000,携带率为1/40~1/50。SMA由运动神经元存活(survive motor neuron, SMN)基因丧失功能突变致病,人的SMN在每条5号染色体上均有2个高度同源且编码相同蛋白的拷贝,即端粒端的SMN1和着丝粒端的SMN2 ,两者只相差5个碱基,其中编码序列仅相差1个碱基, 即7号外显子的第6个碱基C/T(840 C/T)。由于C的改变,使SMN2产生了选择性剪切,转录产物中90%缺少7号外显子,翻译成截短且不稳定的SMN蛋白。临床上绝大部分SMA患者是由于SMN1基因大片段缺失或微小突变导致,而几乎全部SMA病人都含有至少一个SMN2拷贝,因此SMN2 一直是 SMA治疗的理想靶标。
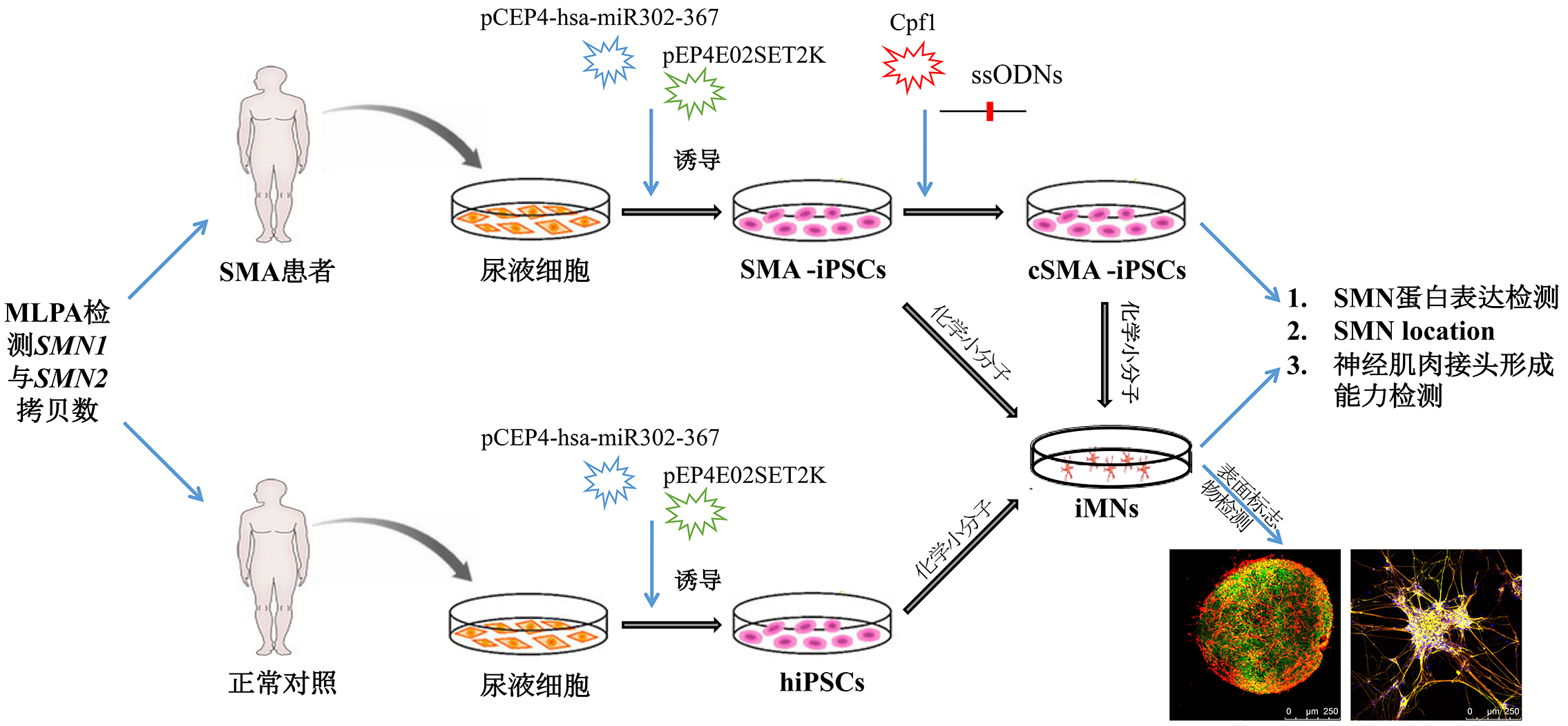
SMA缺乏有效治疗。早期的研究主要通过小分子药物、双功能寡核苷酸、反义寡核苷酸等策略提高全长SMN的mRNA产量,比如2016年FDA批准上市的Spinraza。然而这些方法均需反复给药,不能实现SMN表达的永久恢复。最近AAV介导的SMN1基因替代疗法也进入了临床试验,但仍然存在病毒的安全性和免疫原性等潜在风险。近期有研究利用单链寡聚脱氧核糖核酸(single-stranded oligodeoxynucleotides, ssODNs)将SMN2 840T诱变为C产生类似SMN1基因恢复SMN表达,然而单纯使用ssODNs的效率很低。中南大学梁德生团队采用c-Myc-free非病毒非整合方式将SMA病人的尿液细胞重编程为iPSCs,显著降低诱导重编程的致瘤风险。然后联合高特异性的CRISPR/Cpf1与ssODNs将SMN2 840 T诱变为C,修复效率高达11%。基因修正的iPSCs及其经化学小分子定向分化的运动神经元都恢复了SMN表达和正常分布,并改善了运动神经元形成神经肌肉接头的能力。这是CRISPR/Cpf1应用于病人特异性iPSCs基因编辑治疗研究的首次报道。基因转换策略类似于原位修复,是最理想的基因治疗,本研究的高效率和安全性也为其它遗传病的基因治疗提供了新途径。
《Human Gene Therapy》由欧洲、英国、法国和德国等9个细胞与基因治疗学会联合主办,是该领域最高水平的杂志之一。梁德生教授团队在遗传病的自体化靶向基因治疗研究方面取得了系列原创成果,其中“基因修正血友病A病人iPSCs来源的ECs治疗研究”获法国国家血浆制品集团(LFB)资助进行FDA临床前效果评估。
发表回复
要发表评论,您必须先登录。